What's your position and where are you working?
For the past 5 years I have worked as a lecturer in Biological Chemistry in the section of Biomolecular Medicine at Imperial College London.
Where have you been working before?
Before this, I post-doc’ed at Cambridge; before that, UC Davis; and before that, for my first post-doc, at Imperial College again.
Briefly describe your research.
I am interested in metabolism in invertebrate and microbial species, and how this is involved in several different biological questions. Some of the projects I currently work on include microbial virulence and pathogenesis; how metabolism is affected by problems with recombinant protein folding in the bioprocessing yeast Pichia pastoris; and using earthworms as biomonitors of environmental pollution.
What do you use NMR for?
Together with mass spectrometry, I use NMR for metabolite profiling, as part of the technology for metabolomic studies. Although it’s generally less sensitive than mass spec, NMR still has a very useful role – as a near-universal and robust detector, it can give a quick and information-rich spectral profile. Most of the work we do, we just use 1D NMR for profiling; particularly useful for studies where you want to process as many samples as possible. However there are also other cases where more in-depth NMR experiments are needed, for example for isotopomer analysis to investigate metabolic fluxes; or to assign novel metabolites (essentially a natural products chemistry problem).
Which NMR software are you using?
XWIN-NMR and Topspin for data acquisition; iNMR for all NMR processing. I also use Chenomx NMR Suite for helping assign and quantitate metabolites in NMR spectra.
Which other NMR software have you used in the past?
I’ve also used VNMR and ACDLabs NMR software, and MestreC (before it was released as a commercial product).
How do you rate iNMR?
iNMR is not only my favourite NMR software for Mac OS (I’m not sure how many competitors there are at the moment), but it’s easily my favourite NMR software full stop. It’s one of a handful of Mac-only packages that I use all the time as part of my regular working day (others include Papers, Aabel, and Bookends). Features that I particularly like include the Overlay Manager (which makes it by far the quickest and easiest NMR software to use for comparing multiple spectra, in my opinion), and also the overall simplicity of using it to produce quality spectral images that can go straight into a paper or presentation without having to use multiple machines or virtualization. I admit I wasn’t really bothered by lack of anti-aliasing on spectra before using iNMR, but now I’m used to it, I do find it genuinely annoys me when looking at spectra in Topspin, say – it’s distinctly harder to see fine detail without zooming in. It’s also elegant and quick – not essential properties for software, but makes it more enjoyable to use on a regular basis.
Is it enough for your needs?
Well, it’s certainly enough for my needs in the sense that all of the spectra that I acquire are processed with iNMR – so in one sense, yes, almost by definition. It’s not 100% perfect though, there are still some small issues that could be ironed out in future releases – and as I’ve already said, I do use Chenomx software for some complementary uses which iNMR isn’t primarily designed for. I definitely see it as a crucial part of my workflow for the foreseeable future though, and expect it will keep improving (although by now it’s a relatively mature product).
Wednesday, 2 December 2009
Hands on 3-D Processing
A measure of the computing power available today with a desktop computer is the possibility of processing huge spectra in real time. For example: is it possible to correct the phase of a 3-D matrix interactively, in real time? The answer is: yes and you don't even have to employ more than a single core nor to buy an high-end graphic card. When I mean interactive processing I mean that:
- you see a graphic representation of the matrix at each processing stage.
- you can play with it, for example change a parameter just to see the effect it has on the matrix.
It is always necessary to know and understand the mathematical rules that govern NMR processing. The more you know them the more you enjoy their visual representations. The more you play with the graphics, the more you understand the maths behind. The two things go together.
You can find a self-teaching course on basic 3-D processing on the iNMR web site. There is no theory, only a lot of examples and a good measure of practical tips.
- you see a graphic representation of the matrix at each processing stage.
- you can play with it, for example change a parameter just to see the effect it has on the matrix.
It is always necessary to know and understand the mathematical rules that govern NMR processing. The more you know them the more you enjoy their visual representations. The more you play with the graphics, the more you understand the maths behind. The two things go together.
You can find a self-teaching course on basic 3-D processing on the iNMR web site. There is no theory, only a lot of examples and a good measure of practical tips.
Thursday, 26 November 2009
Open Source NMR freeware
Most of the readers arrive here using Google, without knowing me and my blog. Usually they get very angry because they arrive... on the trapping post I wrote 3 years ago! I want to do something to keep them glad...
So you want "open source" stuff? Do you know what it really means? Are you ready to compile, test, debug it and add a graphic interface to it?
Just because you asked for it, here is a list of available projects. If you know other links, add them into a comment.
CCPN
NPK
matNMR
ProSpectND
Connjur
Newton-NMR
nmrproc
DOSY Toolbox
list of 30+ projects
So you want "open source" stuff? Do you know what it really means? Are you ready to compile, test, debug it and add a graphic interface to it?
Just because you asked for it, here is a list of available projects. If you know other links, add them into a comment.
CCPN
NPK
matNMR
ProSpectND
Connjur
Newton-NMR
nmrproc
DOSY Toolbox
list of 30+ projects
Monday, 16 November 2009
Tips
It rarely happens to find valuable tips about processing on the web. When it happens, it's probably not enough to bookmark the page (it may disappear), copying it is a better idea. Here is the link:
http://spin.niddk.nih.gov/NMRPipe/embo/
When you arrive there, scroll down until you find the chapter Some General Tips About Spectral Processing.
The focus is on multi-dimensional processing with NMRPipe, but a few concepts are generally applicable indeed.
The brief discussion about first-point pre-multiplication is something to bear in mind. You can also find clearly expressed opinions on zero-filling, linear prediction and baseline correction.
http://spin.niddk.nih.gov/NMRPipe/embo/
When you arrive there, scroll down until you find the chapter Some General Tips About Spectral Processing.
The focus is on multi-dimensional processing with NMRPipe, but a few concepts are generally applicable indeed.
The brief discussion about first-point pre-multiplication is something to bear in mind. You can also find clearly expressed opinions on zero-filling, linear prediction and baseline correction.
Saturday, 14 November 2009
Shadow

Here is a picture I have found on the internet. I have never been in this place, if this is what you want to hear. Suppose, instead, that I live just in front of this tower and tomorrow I take a photo of it immediately after dawn, then another photo after 25 hours and so on for a week, with regular intervals of 25h between pictures. Eventually I print all the pictures in order and ask you:
"This pictures have been taken in this exact order at regular intervals. Can you tell me how long the intervals were?".
Somebody will answer: "1 hour" and the answer would be partially correct. A more correct answer would be 1 + 24 n hours, with n = 0, 1, 2, 3….
The world of FT-NMR is similar. The difference is that the regular interval between consecutive observation is known in advance while the speed of the hands and of the shadow is unknown. In other words, it is the opposite of my example of the tower.
The equivalent of a day, in the world of FT-NMR, is very very short and is called dwell time. It forms a Fourier pair, so to speak, with the spectral width. The spectroscopist sets the former, the latter is a mere mathematical consequence.
The ignorant says that the spectral width is the distance from the first point of the spectrum to the last one. This statement is as incorrect as saying that a day is made of 23 hours! The spectral width, actually, is the distance from the first point to the first point after the last one!
Let's verify it with a numerical example. Let's say we have a spectrum of 1024 points, separated by 2 Hz. If we zero-fill the FID up to 2048 points, the distance should decreases to 1 Hz. The spectral width is 2048 Hz in both cases, and this is OK. If you measure it the other way, then you have a spectral width of 2046 Hz that grows up to 2047 Hz. This is absurd, because the value is fixed at acquisition time and can't be changed by processing.
The relation is Spectral_width = 1 / Dwell_time.
Many books report another formula: Spectral_width = 1 / (2 * Dwell_time). This assumes an instrument without quadrature detection, in other words with a single detector. I have never used such an instrument.
To be exact, my whole description is dated, because today's instruments work in oversampling, the actual dwell times are shorter than what the spectroscopist sets (4 or 8 times the value reported), the FID that we see is already the result of a couple of FT (first direct, then inverse), et cetera. Seems complicated but it is not. We see what we need to see, the complications are hidden.
The important concept to remember is that the components of a digital spectrum, even if they are called "points", should be treated and conceptualized as tiles. There is no room in between. This idea will help you when you'll try to save a spectrum as a table of intensity vs. frequency. You will get the correct frequency value of each "point".
Wednesday, 21 October 2009
Trigonometry for Dummies

This is a doublet of doublets. If you move one doublet towards the other, for example by increasing the smaller J, the central peaks will coalesce into a single peak of double intensity (the algebraic sum of the central peaks).

A triplet is a special case of doublet of doublets (the Js are identical).

Another doublet of doublets. This time the smaller coupling is anti-phase.
If we increase the small J, we'll see another algebraic addition.

This is a triplet. Right?
Tuesday, 20 October 2009
NMR for Dummies
Here is where the real fun begins. I was not joking with my previous post. You can really do these things at home. The simple instructions are available on another site, and they are illustrated. After completing the tutorial (it takes 5 minutes in total) a working application will remain in your hard disc, and you will be free to apply the same treatment to your own spectra.
iNMR has always been available as a free download (since 2005). Access is unlimited and the program never expires. Other specialized simulation modules are included to cover most of the needs of an advanced spectroscopist (the few exceptions are motivated by the fact that other needs were already covered by existing freeware).
My blog is three years old. Half of the comments have been dedicated to a single post, which is clearly (and purposely) the least representative of the blog. When people find something useful (and this is certainly the case today) and free, they don't comment.
If you are not going to comment, I will comment by myself.
COMMENT: although it's a serious work, although it contains a lot of maths (and maybe just for this reason) it's funny too.
iNMR has always been available as a free download (since 2005). Access is unlimited and the program never expires. Other specialized simulation modules are included to cover most of the needs of an advanced spectroscopist (the few exceptions are motivated by the fact that other needs were already covered by existing freeware).
My blog is three years old. Half of the comments have been dedicated to a single post, which is clearly (and purposely) the least representative of the blog. When people find something useful (and this is certainly the case today) and free, they don't comment.
If you are not going to comment, I will comment by myself.
COMMENT: although it's a serious work, although it contains a lot of maths (and maybe just for this reason) it's funny too.
Friday, 16 October 2009
Try this at home
Returning to the DQF-COSY of taxole, I have found this challenge:

I couldn't tell which kind of multiplet it was. I have extracted the section corresponding to the red line and moved it into my novel simulator. Then I have introduced 3 couplings and pushed the "Fit" button.

That's all. The goodness of the fit is convincing. It is a doublet of doublets of doublets and the Js are: 14.7, 6.4 and 9.9 Hz. The latter corresponds to an antiphase coupling. Not only I know which kind of multiplet it is, I have also measured the Js!
You might feel it's funny, I find it amazing, perhaps somebody will say it is useful. "The coupling constants were extracted from the DQF-COSY by the blogger's simulator".

I couldn't tell which kind of multiplet it was. I have extracted the section corresponding to the red line and moved it into my novel simulator. Then I have introduced 3 couplings and pushed the "Fit" button.

That's all. The goodness of the fit is convincing. It is a doublet of doublets of doublets and the Js are: 14.7, 6.4 and 9.9 Hz. The latter corresponds to an antiphase coupling. Not only I know which kind of multiplet it is, I have also measured the Js!
You might feel it's funny, I find it amazing, perhaps somebody will say it is useful. "The coupling constants were extracted from the DQF-COSY by the blogger's simulator".
Digitization
I am continuing my explorations on board of my new multiplet simulator. It has been easy to simulate an asymmetric doublet. Here the asymmetry is only apparent and is due to the limited digitization.

As explained yesterday, the black line belongs to an experimental 2-D spectrum (today I have chosen a TOCSY), the red line is a theoretical doublet acquired and processed under the same conditions.
My simulator allows me to change the frequency by dragging the blue label at the bottom.

In this way the coupling remains constant. It turns out that my spectrum was a middle-range case. With a slight increase of the chemical shift I can obtain a symmetric doublet, or even a singlet!


As explained yesterday, the black line belongs to an experimental 2-D spectrum (today I have chosen a TOCSY), the red line is a theoretical doublet acquired and processed under the same conditions.
My simulator allows me to change the frequency by dragging the blue label at the bottom.

In this way the coupling remains constant. It turns out that my spectrum was a middle-range case. With a slight increase of the chemical shift I can obtain a symmetric doublet, or even a singlet!

Thursday, 15 October 2009
Natural Products

A reader asked me: "What is the shape of a 2-D peak?" and my answer was: "Exponential! (in time domain)". This is all you need to know, I think. The picture should help, as always. If you are reading this blog, it means that you have something like a computer. Even the iPhone is a computer (you can run programs on it) and can display my blog. I suspect, however, that you have a better computer, like I do.
You can use your computer to run simulations and more: you can ask it to fit a model spectrum to an experimental spectrum. This is what I have done. The black line is a (fragment of a projection of a) DQF-COSY. The red line is a model generated by the computer. All I said was:
1) the signal has an exponential decay;
2) it's an anti-phase doublet;
3) here is an experimental spectrum and here are its processing parameters;
4) please fit the model to my spectrum!
The goal was a better estimate of the coupling constant. The distance between the two peaks is 7.3 Hz, while the model contains a J = 8.88 Hz. My impression is that the model is more accurate by 1.6 Hz.
The calculation involved is very little (the whole process was faster than the blinking of an eye). The job of programming was more time-consuming. If you don't like spending time, find someone else that can do the job for you. They ask 0.99$ (0.79€) for an application (for the iPhone). Calculating the Js with more accuracy is certainly worth the price.
There is no secret, no new theory, should you need more explanations please ask.
If you want to know more, I can try to explain what was behind the question of the reader. It's not necessary, it's an un-necessary complication, but it's also well-know theory. If you submit to FT a function (e.g.: an exponential decay) you get a different function (in the example, a Lorentzian). They form a "Fourier pair". Being that most 2-D spectra are weighted with a squared cosine-bell my friend probably wanted to know which other function it makes a pair with.
Now you can realize that it is more a theoretical question than a practical one: if the final goal is to simulate an NMR spectrum, it's enough to know that the signal decays exponentially (and everybody knows it).
The program shown by the picture can simulate a multiplet (with 3 different J values). The multiplicities that can be chosen from the menus are:

but it's trivial to extent the menu and handle quintets and so on. Even without further modifications, you can already simulate a quartet of triplets of doublets."R" is the inverse of the transversal relaxation time. This kind of program can't simulate second-order spectra, but nothing prevents you from writing a program that simulates second-order spectra in time domain.
What happens when the user pushes the "Fit" button? The computer runs a Levenberg-Marquardt optimization, based on first derivatives. How have I found the derivatives without knowing the function that describes a peak? You know: life is simpler when you have a computer...
Monday, 28 September 2009
interviews
Here is the complete list of my interviews:
Kevin Theisen
Stefano Antoniutti
Arthur Roberts
Antonio Randazzo
Bernhard Jaun
Daniel J. Weix
Jacek Stawinski
Stefano Ciurli
Jake Bundy
If you like to be interviewed, please send both questions and answers via email (or as a comment here).
The next Euromar conference will be held in Florence from July 4 to July 9 2010. I have no intention to attend but I want to be there. If you plan to attend, contact me via email or phone and let's arrange a meeting. I would be delighted to know you in person.
Kevin Theisen
Stefano Antoniutti
Arthur Roberts
Antonio Randazzo
Bernhard Jaun
Daniel J. Weix
Jacek Stawinski
Stefano Ciurli
Jake Bundy
If you like to be interviewed, please send both questions and answers via email (or as a comment here).
The next Euromar conference will be held in Florence from July 4 to July 9 2010. I have no intention to attend but I want to be there. If you plan to attend, contact me via email or phone and let's arrange a meeting. I would be delighted to know you in person.
Tuesday, 18 August 2009
Stefano Ciurli
Q. Please introduce yourself to the readers of the blog.
A. I am a full professor of general and inorganic chemistry at the University of Bologna (Italy).
I received my M.Sc. from the University of Pisa with a thesis done at Columbia University (NY), received my Ph.D. in chemistry from Harvard University, and done a post-doctoral work at the NMR center in Florence.
I work on the structural biology and biological chemistry of metalloproteins. After several years of work on electron transfer proteins containing Fe and Cu, I have been spending the last ten years working on the biochemistry of nickel.
I use NMR mainly for processing and visualization of 1D, 2D, and 3D NMR spectra of proteins prior to go on and use other programs for more dedicated tasks of spectral signal assignment.
Q. Which NMR software are you using?
A. The first choice is iNMR, for its amazing speed and flexibility, especially for 3D spectra. Easy to use and great performance. I use MestreNova for teaching purposes (mainly because of lack of Mac computers among the students, otherwise that would be perfect, considering the simulation modules etc.), and NMRPipe for other different tasks more dedicated to protein NMR.
Q. Is iNMR enough for your needs?
A. I like to use both iNMR and NMRPipe, having iNMR as the first choice and leaving NMRPipe for more specialized work.
A. I am a full professor of general and inorganic chemistry at the University of Bologna (Italy).
I received my M.Sc. from the University of Pisa with a thesis done at Columbia University (NY), received my Ph.D. in chemistry from Harvard University, and done a post-doctoral work at the NMR center in Florence.
I work on the structural biology and biological chemistry of metalloproteins. After several years of work on electron transfer proteins containing Fe and Cu, I have been spending the last ten years working on the biochemistry of nickel.
I use NMR mainly for processing and visualization of 1D, 2D, and 3D NMR spectra of proteins prior to go on and use other programs for more dedicated tasks of spectral signal assignment.
Q. Which NMR software are you using?
A. The first choice is iNMR, for its amazing speed and flexibility, especially for 3D spectra. Easy to use and great performance. I use MestreNova for teaching purposes (mainly because of lack of Mac computers among the students, otherwise that would be perfect, considering the simulation modules etc.), and NMRPipe for other different tasks more dedicated to protein NMR.
Q. Is iNMR enough for your needs?
A. I like to use both iNMR and NMRPipe, having iNMR as the first choice and leaving NMRPipe for more specialized work.
Friday, 24 July 2009
QR-DOSY
At the beginning of the month we saw that there are cases where the diffusion coefficients can be measured with the same mathematical tools used to measure relaxation. We have seen DOSY spectra of pure compounds where each signal decays as a pure exponential. Even a mixture can behave in the same way, if the signals don't overlap. In summary, calculating the diffusion coefficients is often easy.
What I was curious to discover was: is it possible to recalculate the components of a mixture if the diffusion coeffients are known? From a pure mathematical point of view the answer was already: "yes", but I wanted to verify it in practice.
As far as I know, something like: "QR-DOSY" has never been mentioned. So many DOSY methods already exist and I don't mind to increase the Babel with yet another acronym. Do you?
THEORY
If we know the diffusion coefficient D(j) we can calculate that the NMR signal in the spectrum i will be proportional to a value A(i,j) = exp(-D(j)F(i)) where F is a function of the gyromagnetic ratio, the gradient strength, the diffusion delay, etc.. but not a function of the chemical shift nor of the diffusion coefficient.
For each column of the DOSY spectrum we have a system of equations: Ax = b.
x = intensity of the spectrum of the pure compounds at the chemical shift that coresponds to the given column.
b = intensity of the DOSY along the same column.
We know A and b, therefore we can calculate x. A is the same for all the columns and this is a great advantage. We can apply a well known decomposition: Rx = Qb.
Calculating Q and R from A takes time, but we need to do it only once. Then then computer can swiftly solve all the systems in the form Rx = Qb.
METHODS
Obviously, we will find that some values of the x will be negative. In such a case, we can choose a subset of A (omitting the component with negative intensity) and solve the reduced problem. This simplifying mechanism can be applied iteratively, even when the value of x is positive yet small.
RESULTS

This is the same old spectrum we are familiar with, processed with the new QR-DOSY.
DISCUSSION
Two components are completely separated. The third component is not, although at this point it becomes easy to recognize its peaks. Other cases I have studied yield similar results, maybe not as nice. Advantages of the QR-DOSY method:
- easy to understand;
- easy to use WITH THE ASSISTANCE of a software for the book-keeping activity (like measuring the diffusion coefficients);
- fast;
- the user can play with a few parameters, trying to improve the results;
- the final spectra are clean from artifacts.
Cons:
- not all the components are always resolved;
- it's a problem if two diffusion coefficients are similar (of course the program itself can easily detect this circumstance).
What I was curious to discover was: is it possible to recalculate the components of a mixture if the diffusion coeffients are known? From a pure mathematical point of view the answer was already: "yes", but I wanted to verify it in practice.
As far as I know, something like: "QR-DOSY" has never been mentioned. So many DOSY methods already exist and I don't mind to increase the Babel with yet another acronym. Do you?
THEORY
If we know the diffusion coefficient D(j) we can calculate that the NMR signal in the spectrum i will be proportional to a value A(i,j) = exp(-D(j)F(i)) where F is a function of the gyromagnetic ratio, the gradient strength, the diffusion delay, etc.. but not a function of the chemical shift nor of the diffusion coefficient.
For each column of the DOSY spectrum we have a system of equations: Ax = b.
x = intensity of the spectrum of the pure compounds at the chemical shift that coresponds to the given column.
b = intensity of the DOSY along the same column.
We know A and b, therefore we can calculate x. A is the same for all the columns and this is a great advantage. We can apply a well known decomposition: Rx = Qb.
Calculating Q and R from A takes time, but we need to do it only once. Then then computer can swiftly solve all the systems in the form Rx = Qb.
METHODS
Obviously, we will find that some values of the x will be negative. In such a case, we can choose a subset of A (omitting the component with negative intensity) and solve the reduced problem. This simplifying mechanism can be applied iteratively, even when the value of x is positive yet small.
RESULTS

This is the same old spectrum we are familiar with, processed with the new QR-DOSY.
DISCUSSION
Two components are completely separated. The third component is not, although at this point it becomes easy to recognize its peaks. Other cases I have studied yield similar results, maybe not as nice. Advantages of the QR-DOSY method:
- easy to understand;
- easy to use WITH THE ASSISTANCE of a software for the book-keeping activity (like measuring the diffusion coefficients);
- fast;
- the user can play with a few parameters, trying to improve the results;
- the final spectra are clean from artifacts.
Cons:
- not all the components are always resolved;
- it's a problem if two diffusion coefficients are similar (of course the program itself can easily detect this circumstance).
Tuesday, 14 July 2009
Jacek Stawinski
Q. What's your position and where are you working?
A. I am Professor of Organic Chemistry at Stockholm University, Stockholm, Sweden, and at the Institute of Bioorganic Chemistry, Polish Academy of Science, Poznan, Poland.
Q. Where have you been working before?
A. Adam Mickiewicz University, Poznan, Poland, and Institute of Organic Chemistry, Polish Academy of Science, Warsaw, Poland
Q. Briefly describe your research.
A. My field of expertise is bioorganic phosphorus chemistry, nucleic acids chemistry, and lipid and phospholipid chemistry (http://www.organ.su.se/js).
Q. What do you use NMR for?
A. Characterization of synthetic intermediates, structure determination, spin simulations, NMR dynamic processes.
Q. Which NMR software are you using now?
iNMR, the latest version.
Q. Which other NMR software have you used in the past?
A. Swan NMR, Topspin, MestreC, MNOVA
Q. How do you rate iNMR?
A. iNMR is superior, by far, to all NMR software I have used. It provides a powerful, intuitive and professional environment for processing and plotting NMR data. The software is very fast, has scripting ability, and a lot of keyboard shortcuts and useful extras. No doubts, iNMR is right on the cutting edge on the NMR processing software development. Due to simple interface, iNMR is a user friendly application, but it hides a lot of powerful tools for advanced tasks. And last, but not least, the support from the programmer is prompt, competent, and friendly.
Q. Is it enough for your needs?
A. I have never faced a situation when iNMR could not do, what other software can. For me, it is the tool of choice for dynamic NMR. Also very useful during teaching NMR courses.
A. I am Professor of Organic Chemistry at Stockholm University, Stockholm, Sweden, and at the Institute of Bioorganic Chemistry, Polish Academy of Science, Poznan, Poland.
Q. Where have you been working before?
A. Adam Mickiewicz University, Poznan, Poland, and Institute of Organic Chemistry, Polish Academy of Science, Warsaw, Poland
Q. Briefly describe your research.
A. My field of expertise is bioorganic phosphorus chemistry, nucleic acids chemistry, and lipid and phospholipid chemistry (http://www.organ.su.se/js).
Q. What do you use NMR for?
A. Characterization of synthetic intermediates, structure determination, spin simulations, NMR dynamic processes.
Q. Which NMR software are you using now?
iNMR, the latest version.
Q. Which other NMR software have you used in the past?
A. Swan NMR, Topspin, MestreC, MNOVA
Q. How do you rate iNMR?
A. iNMR is superior, by far, to all NMR software I have used. It provides a powerful, intuitive and professional environment for processing and plotting NMR data. The software is very fast, has scripting ability, and a lot of keyboard shortcuts and useful extras. No doubts, iNMR is right on the cutting edge on the NMR processing software development. Due to simple interface, iNMR is a user friendly application, but it hides a lot of powerful tools for advanced tasks. And last, but not least, the support from the programmer is prompt, competent, and friendly.
Q. Is it enough for your needs?
A. I have never faced a situation when iNMR could not do, what other software can. For me, it is the tool of choice for dynamic NMR. Also very useful during teaching NMR courses.
Thursday, 9 July 2009
Bull's-Eyes
Here are some caffeine peaks (below) and EthoxyEthanol peaks (above). The display is DOSY-like, yet the numerical treatment is a simpler and faster mono-exponential fit. Each column has been processed independently from the rest, and each column yields a different result:

Now let's apply a Whittaker Smoother, with a small value of lambda (100), along each ROW:

or a big lambda (500):

or a huge value (8000):

The smoother averages the results obtained from the different columns. The peaks are perfectly aligned.

Now let's apply a Whittaker Smoother, with a small value of lambda (100), along each ROW:

or a big lambda (500):

or a huge value (8000):

The smoother averages the results obtained from the different columns. The peaks are perfectly aligned.
Wandering
Jean Marc Nuzillard writes:
I agree with the idea. Actually I had arrived at the same conclusion for a different reason. Look at this spectrum (a DOSY after FT):
 The signal/noise ratio is low yet acceptable, the compound is pure, the decay mono-exponential. No concern about phase and baseline. Alas, this peak is too nasty for my tastes. The frequency is not constant over time. The "trivial" trick by Jean-Marc should work. I give my preference to binning, because the output of binning is not a numerical table (like with integration) but a new spectrum, so the same NMR program can be used for the subsequent exponential fit.
The signal/noise ratio is low yet acceptable, the compound is pure, the decay mono-exponential. No concern about phase and baseline. Alas, this peak is too nasty for my tastes. The frequency is not constant over time. The "trivial" trick by Jean-Marc should work. I give my preference to binning, because the output of binning is not a numerical table (like with integration) but a new spectrum, so the same NMR program can be used for the subsequent exponential fit.
Before Carlos corrects me, let me stress a few details. We have processed the same experiment with two different algorithms. I got the butterflies and I am not proud of it. Carlos' pictures are a little smaller and cannot be compared:
 (taken from his blog). Anyway, we are really confusing the matter here. From what I understand, the purpose of Carlos was to show how Bayesian DOSY can effectively separate the components of a mixture. I can't express any opinion, because as I said I only have two experiments to work with. In both cases there is no superposition of peaks, therefore there is nothing to separate. I normally like simple examples like these, yet I acknowledge they are not enough.
(taken from his blog). Anyway, we are really confusing the matter here. From what I understand, the purpose of Carlos was to show how Bayesian DOSY can effectively separate the components of a mixture. I can't express any opinion, because as I said I only have two experiments to work with. In both cases there is no superposition of peaks, therefore there is nothing to separate. I normally like simple examples like these, yet I acknowledge they are not enough.
Today I could repeat the same processing of Carlos, because he himself has very kindly given me both the raw data and the software. But 1) I am not terribly interested into this comparison 2) He doesn't give me these things for free for me to criticize him 3) If I really want to do such a thing I'll post a comment directly on his blog.
Now let's go on: ho to make the butterflies go away? The next post shows how to transform a butterfly into bull's-eyes.
The example that is provided by Carlos in his blog would benefit from a processing trick I use when there is no multiplet superimposition:
I simply integrate the multiplets that are recorded at different gradient intensities and I perform a monoexponential fit on integral values.
I suspect the noise on integral values is lower than the one in individual columns of the 1D spectra set, thus making D values more accurate.
Bye bye butterflies. This idea is absolutely trivial but maybe it would be interesting
to implement it.
I agree with the idea. Actually I had arrived at the same conclusion for a different reason. Look at this spectrum (a DOSY after FT):
 The signal/noise ratio is low yet acceptable, the compound is pure, the decay mono-exponential. No concern about phase and baseline. Alas, this peak is too nasty for my tastes. The frequency is not constant over time. The "trivial" trick by Jean-Marc should work. I give my preference to binning, because the output of binning is not a numerical table (like with integration) but a new spectrum, so the same NMR program can be used for the subsequent exponential fit.
The signal/noise ratio is low yet acceptable, the compound is pure, the decay mono-exponential. No concern about phase and baseline. Alas, this peak is too nasty for my tastes. The frequency is not constant over time. The "trivial" trick by Jean-Marc should work. I give my preference to binning, because the output of binning is not a numerical table (like with integration) but a new spectrum, so the same NMR program can be used for the subsequent exponential fit. Before Carlos corrects me, let me stress a few details. We have processed the same experiment with two different algorithms. I got the butterflies and I am not proud of it. Carlos' pictures are a little smaller and cannot be compared:
 (taken from his blog). Anyway, we are really confusing the matter here. From what I understand, the purpose of Carlos was to show how Bayesian DOSY can effectively separate the components of a mixture. I can't express any opinion, because as I said I only have two experiments to work with. In both cases there is no superposition of peaks, therefore there is nothing to separate. I normally like simple examples like these, yet I acknowledge they are not enough.
(taken from his blog). Anyway, we are really confusing the matter here. From what I understand, the purpose of Carlos was to show how Bayesian DOSY can effectively separate the components of a mixture. I can't express any opinion, because as I said I only have two experiments to work with. In both cases there is no superposition of peaks, therefore there is nothing to separate. I normally like simple examples like these, yet I acknowledge they are not enough.Today I could repeat the same processing of Carlos, because he himself has very kindly given me both the raw data and the software. But 1) I am not terribly interested into this comparison 2) He doesn't give me these things for free for me to criticize him 3) If I really want to do such a thing I'll post a comment directly on his blog.
Now let's go on: ho to make the butterflies go away? The next post shows how to transform a butterfly into bull's-eyes.
Pictures
In my fourth year of blogging I have started publishing pictures of spectra. I like my pictures, yet this is not the point. I don't want to convince you that my pictures are beautiful. I want to convince you that I am a spectroscopist and not a programmer.
The programmers have always stated that "in the near future" it will be possible to obtain a completely automatic analysis: from the sample directly to the response (meaning a chemical formula or a list of values), by-passing the plotted spectrum. I have found the same concept and expression "near future", scattered in the literature of all the decades, from the 60s onward.
I really believe it: someday in the future we'll arrive at the completely automatic analysis.
Being that, at this writing time, I am working as a programmer and not as a spectroscopist, I should adhere to this belief and be happy. It happens, instead, that I always think like a spectroscopist scared of remaining unemployed. I really hate to design and write programs for automatic processing and reporting. I like creating programs to display the spectra.
Here comes the difference between "diffusion" and "DOSY". The mere idea of DOSY is to make a troble-free program: push this button and you'll have everything, the diffusion coefficients and the individual components of the mixture. This is what I have understood up to now. In my whole life I have only worked with 2 DOSY experiments, which have not been carried out by me. I have already shown the first one and today I am going to show the second one (please wait a few minutes..).
The programmers have always stated that "in the near future" it will be possible to obtain a completely automatic analysis: from the sample directly to the response (meaning a chemical formula or a list of values), by-passing the plotted spectrum. I have found the same concept and expression "near future", scattered in the literature of all the decades, from the 60s onward.
I really believe it: someday in the future we'll arrive at the completely automatic analysis.
Being that, at this writing time, I am working as a programmer and not as a spectroscopist, I should adhere to this belief and be happy. It happens, instead, that I always think like a spectroscopist scared of remaining unemployed. I really hate to design and write programs for automatic processing and reporting. I like creating programs to display the spectra.
Here comes the difference between "diffusion" and "DOSY". The mere idea of DOSY is to make a troble-free program: push this button and you'll have everything, the diffusion coefficients and the individual components of the mixture. This is what I have understood up to now. In my whole life I have only worked with 2 DOSY experiments, which have not been carried out by me. I have already shown the first one and today I am going to show the second one (please wait a few minutes..).
Saturday, 4 July 2009
Ghost of a Butterfly

Sometimes the butterfly disappears when the picture is scaled down, like into this web page. Click it!
Butterflies
Carlos kindly gave me a copy of the DOSY spectrum he showed on his blog last year.
I am experimenting alternative processing routes. Here is a detail of the caffeine peaks, after applying the rudest (and probably simplest) treatment. The decays have been linearized (by taking the logarithm). The slope of the line is proportional to the diffusion coefficient. The final results are reported as a normal DOSY spectrum.
Click on the thumbnail to see the image at natural scale.

There is less signal/noise in the tails of the peaks, obviously, therefore the error increases: graphically we see the wings of a butterfly.
Being that it's impossible to correct the phase perfectly, some butterflies are asymmetric.
I am experimenting alternative processing routes. Here is a detail of the caffeine peaks, after applying the rudest (and probably simplest) treatment. The decays have been linearized (by taking the logarithm). The slope of the line is proportional to the diffusion coefficient. The final results are reported as a normal DOSY spectrum.
Click on the thumbnail to see the image at natural scale.

There is less signal/noise in the tails of the peaks, obviously, therefore the error increases: graphically we see the wings of a butterfly.
Being that it's impossible to correct the phase perfectly, some butterflies are asymmetric.
Friday, 12 June 2009
Daniel J. Weix
Q. What's your position and where are you working?
A. Assistant Professor, University of Rochester.
Q. Briefly describe your research.
A. Synthetic organic methodology, especially catalysis.
Q. What do you use NMR for?
A. Assaying purity of synthesized materials and identification of products (both organic and inorganic).
Q. Which NMR software are you using?
A. We use iNMR, latest version.
Q. Which other NMR software have you used in the past?
A. I have used MestreC, Bruker XWIN-PLOT, NUTS, MacNUTS (new version), and MestreNOVA.
Q. How do you rate iNMR?
A. I like iNMR better than all of the other software that I have used. The workflow is good and I'm already almost as fast as I was under NUTS (the fastest software of those mentioned above). It seems superior to MestreNOVA, with better keyboard shortcuts (MestreNOVA involved a lot of clicking) and a better user interface. New features are discoverable vs. having to dig into the manual. Importantly, my students like the software very much and they can be taught to use it in very short order.
Q. Is it enough for your needs?
A. More important to me than a list of features is the usability of the program. What good are features that sound nice, but are buggy or so round-about to use that they take more time than the result is worth? Can I get the results I need easily? Can we produce publication quality images? Is it fast? Two features that are often overlooked are the quality of the manual and the customer service of the company behind the software. In these areas iNMR excels. For us, iNMR is more than adequate!
A. Assistant Professor, University of Rochester.
Q. Briefly describe your research.
A. Synthetic organic methodology, especially catalysis.
Q. What do you use NMR for?
A. Assaying purity of synthesized materials and identification of products (both organic and inorganic).
Q. Which NMR software are you using?
A. We use iNMR, latest version.
Q. Which other NMR software have you used in the past?
A. I have used MestreC, Bruker XWIN-PLOT, NUTS, MacNUTS (new version), and MestreNOVA.
Q. How do you rate iNMR?
A. I like iNMR better than all of the other software that I have used. The workflow is good and I'm already almost as fast as I was under NUTS (the fastest software of those mentioned above). It seems superior to MestreNOVA, with better keyboard shortcuts (MestreNOVA involved a lot of clicking) and a better user interface. New features are discoverable vs. having to dig into the manual. Importantly, my students like the software very much and they can be taught to use it in very short order.
Q. Is it enough for your needs?
A. More important to me than a list of features is the usability of the program. What good are features that sound nice, but are buggy or so round-about to use that they take more time than the result is worth? Can I get the results I need easily? Can we produce publication quality images? Is it fast? Two features that are often overlooked are the quality of the manual and the customer service of the company behind the software. In these areas iNMR excels. For us, iNMR is more than adequate!
Wednesday, 10 June 2009
Bernhard Jaun
Q. What's your position and where are you working?
A. Professor in Organic Chemistry at ETH Zurich (Swiss Federal Institute of Technology). Head of the NMR labs.
Q. Where have you been working before?
A. Columbia University New York, then ETH Zurich for the last 29 years.
Q. Briefly describe your research.
A. Physical organic chemistry, in particular its application to biological questions.
Q. What do you use NMR for?
A. I am the head of NMR operations in an institute with more than 200 scientists. Most of them use NMR. The applications go from 3D solution structures of biopolymers to physical organic applications of NMR (such as host guest complexation, dynamic processes, thermodynamics and kinetics) to structure elucidation of novel natural compounds and (for the majority) characterization of synthetic intermediates.
Q. Which NMR software are you using now?
A. Topspin, VNMR, Mnova, iNMR, plus specialized software for 3D solution structure calculation such as XPLOR, SPARKY, CNS, DYANA, MARDIGRAS etc.
We have quite a large percentage of people using Macs (ca. 50%) in our institute.
Q. Which other NMR software have you used in the past?
A. SwaNMR for Mac OS9 and most of the software used for NMR over the last thirty years.
Q. How do you rate iNMR?
A. iNMR is a good and fast program which can do practically all of the work an NMR spectroscopist will ever need. Its strength lies in the flexibility and its more mathematical/physical approach to NMR such as beeing able to do all kinds of transforms, simulating dynamic exchange problems, analyzing spin systems etc. Clearly, the program is written by an NMR specialist for NMR spectroscopists.
The "weaknesses" are in the fact that iNMR is not as easy to learn as some other programs by people who do not know much about NMR and are only interested in getting "nice" plots and listings for their synthetic papers etc. Compared to other programs, iNMR uses only a fraction of icons and palettes but insiders can work very efficiently because of all the keyboard shortcuts and the scripting ability. The current versions still have a few bugs or inconveniences in the field of graphics, e.g. when it comes to plot 1D spectra at the border of 2Ds etc., axis adjustments when changing the window size etc. [Editor's note: this interview refers to the old version 3; the current version 4, made with the collaboration of prof. Jaun himself, solved all the above mentioned problems].
Maybe the best point about iNMR is that according to my experience, there is no other software where the programmer is so fast in responding to either bug reports or demands for new features. So, if I still sometimes get angry about a bug (or something I want to do but cant find out how) in iNMR, it is usually my own fault because I did'nt take the time to write to the author about it. If I had contacted the author, the problem would long be solved by now. Compare that to MS Office or the spectrometer manufacturers NMR programs!
Q. Is it enough for your needs?
A. We NMR spectroscopists have to accept that for a majority of the scientists in todays chemistry/biology research, NMR is a black box that's neverless - and "unfortunately"- absolutely necessary. They like to use software that seems to generate listings and plots without requiring knowledge by the operator. We still try to teach our own students about the innards of NMR-experiments. But the reality is that black-box attitude and the trend for automation are increasing all the time.
I think that in my domain of responsability with 200 scientists using NMR, it might actually be a good idea to start to write some scripts for iNMR that do all the standard processing for routine spectra. This might make iNMR more poular for all those, who are not really interested in the inner workings and just need a nice plot to show to their supervisor and who now rather use MNOVA for Mac because they think it is easier to use.
Also, I think that iNMR could become the tool of choice for all special and more physical things that can be done by NMR. In particular, there is only a very limited number of still living programs that can iteratively fit dynamic spectra from complicated exchanging spin systems. Other things I could think of are extracting coupling constants from 2Ds by simulation of cross peaks, analysing relaxation data, measuring residual dipolar couplings from heteronuclear 2D spectra, diffusion etc. etc.
A. Professor in Organic Chemistry at ETH Zurich (Swiss Federal Institute of Technology). Head of the NMR labs.
Q. Where have you been working before?
A. Columbia University New York, then ETH Zurich for the last 29 years.
Q. Briefly describe your research.
A. Physical organic chemistry, in particular its application to biological questions.
Q. What do you use NMR for?
A. I am the head of NMR operations in an institute with more than 200 scientists. Most of them use NMR. The applications go from 3D solution structures of biopolymers to physical organic applications of NMR (such as host guest complexation, dynamic processes, thermodynamics and kinetics) to structure elucidation of novel natural compounds and (for the majority) characterization of synthetic intermediates.
Q. Which NMR software are you using now?
A. Topspin, VNMR, Mnova, iNMR, plus specialized software for 3D solution structure calculation such as XPLOR, SPARKY, CNS, DYANA, MARDIGRAS etc.
We have quite a large percentage of people using Macs (ca. 50%) in our institute.
Q. Which other NMR software have you used in the past?
A. SwaNMR for Mac OS9 and most of the software used for NMR over the last thirty years.
Q. How do you rate iNMR?
A. iNMR is a good and fast program which can do practically all of the work an NMR spectroscopist will ever need. Its strength lies in the flexibility and its more mathematical/physical approach to NMR such as beeing able to do all kinds of transforms, simulating dynamic exchange problems, analyzing spin systems etc. Clearly, the program is written by an NMR specialist for NMR spectroscopists.
The "weaknesses" are in the fact that iNMR is not as easy to learn as some other programs by people who do not know much about NMR and are only interested in getting "nice" plots and listings for their synthetic papers etc. Compared to other programs, iNMR uses only a fraction of icons and palettes but insiders can work very efficiently because of all the keyboard shortcuts and the scripting ability. The current versions still have a few bugs or inconveniences in the field of graphics, e.g. when it comes to plot 1D spectra at the border of 2Ds etc., axis adjustments when changing the window size etc. [Editor's note: this interview refers to the old version 3; the current version 4, made with the collaboration of prof. Jaun himself, solved all the above mentioned problems].
Maybe the best point about iNMR is that according to my experience, there is no other software where the programmer is so fast in responding to either bug reports or demands for new features. So, if I still sometimes get angry about a bug (or something I want to do but cant find out how) in iNMR, it is usually my own fault because I did'nt take the time to write to the author about it. If I had contacted the author, the problem would long be solved by now. Compare that to MS Office or the spectrometer manufacturers NMR programs!
Q. Is it enough for your needs?
A. We NMR spectroscopists have to accept that for a majority of the scientists in todays chemistry/biology research, NMR is a black box that's neverless - and "unfortunately"- absolutely necessary. They like to use software that seems to generate listings and plots without requiring knowledge by the operator. We still try to teach our own students about the innards of NMR-experiments. But the reality is that black-box attitude and the trend for automation are increasing all the time.
I think that in my domain of responsability with 200 scientists using NMR, it might actually be a good idea to start to write some scripts for iNMR that do all the standard processing for routine spectra. This might make iNMR more poular for all those, who are not really interested in the inner workings and just need a nice plot to show to their supervisor and who now rather use MNOVA for Mac because they think it is easier to use.
Also, I think that iNMR could become the tool of choice for all special and more physical things that can be done by NMR. In particular, there is only a very limited number of still living programs that can iteratively fit dynamic spectra from complicated exchanging spin systems. Other things I could think of are extracting coupling constants from 2Ds by simulation of cross peaks, analysing relaxation data, measuring residual dipolar couplings from heteronuclear 2D spectra, diffusion etc. etc.
Tuesday, 9 June 2009
Antonio Randazzo
Q. What's your position and where are you working?
A. I am an Associate Professor at the Faculty of Pharmacy - University of Naples "Federico II"- Italy
Q. Where have you been working before?
A. I have worked also at The Scripps Research Institute (San Diego - California - USA) and at the Vanderbilt University (Nashville - Tennessee - USA)
Q. Briefly describe your research.
A. I have worked in the field of Bioactive Natural Product. I was in charge of the isolation and structural elucidation of new secondary metabolites from marine organisms. The characterization of the new compounds has been accomplished mainly by NMR. Then I moved to the structural study of protein by NMR. Currently I study unusual structures of DNA. In particular I study the structure of modified DNA quadruplex structures....always by means of NMR. I had the occasion to use NMR also in the field of food science.
Q. Which NMR software are you using now?
A. Currently I am using iNMR on two different machines: an iMAC and a brand new MAC PRO both running Leopard OS and both equipped with two monitors. I find really cool to display 2-3-4 spectra distributed between the two screens and using the recently developed "global cross" feature to display a synchronized cursor simultaneously in all spectra. In this way the assignment of whatever molecule become very simple even in the case of complex and overlapped spectra.
Q. Which other NMR software have you used in the past?
A. I have used many NMR softwares. However, I have used extensively Xeasy and Felix (Accelrys, San Diego USA).
Q. How do you rate iNMR?
A. Top score!
Q. Is it enough for your needs?
A. I find it an EXCELLENT software. I am impressed on the very high quality processing features and the very easy way to use it. It is fast and very user friendly. It satisfies completely all my needs and it is also affordable. Furthermore, It is great in commenting the spectra in order to get a nice pictures for scientific work or didactics. Moreover, the after sale assistance to the software is absolutely incomparable with other softwares. Each improvement I have asked for the software, it has been realized in hours!!!! The assistance is the best ever. I have not found anything like the iNMR assistance before in all my carrier. I definitively give to iNMR my strongest recommendation.
A. I am an Associate Professor at the Faculty of Pharmacy - University of Naples "Federico II"- Italy
Q. Where have you been working before?
A. I have worked also at The Scripps Research Institute (San Diego - California - USA) and at the Vanderbilt University (Nashville - Tennessee - USA)
Q. Briefly describe your research.
A. I have worked in the field of Bioactive Natural Product. I was in charge of the isolation and structural elucidation of new secondary metabolites from marine organisms. The characterization of the new compounds has been accomplished mainly by NMR. Then I moved to the structural study of protein by NMR. Currently I study unusual structures of DNA. In particular I study the structure of modified DNA quadruplex structures....always by means of NMR. I had the occasion to use NMR also in the field of food science.
Q. Which NMR software are you using now?
A. Currently I am using iNMR on two different machines: an iMAC and a brand new MAC PRO both running Leopard OS and both equipped with two monitors. I find really cool to display 2-3-4 spectra distributed between the two screens and using the recently developed "global cross" feature to display a synchronized cursor simultaneously in all spectra. In this way the assignment of whatever molecule become very simple even in the case of complex and overlapped spectra.
Q. Which other NMR software have you used in the past?
A. I have used many NMR softwares. However, I have used extensively Xeasy and Felix (Accelrys, San Diego USA).
Q. How do you rate iNMR?
A. Top score!
Q. Is it enough for your needs?
A. I find it an EXCELLENT software. I am impressed on the very high quality processing features and the very easy way to use it. It is fast and very user friendly. It satisfies completely all my needs and it is also affordable. Furthermore, It is great in commenting the spectra in order to get a nice pictures for scientific work or didactics. Moreover, the after sale assistance to the software is absolutely incomparable with other softwares. Each improvement I have asked for the software, it has been realized in hours!!!! The assistance is the best ever. I have not found anything like the iNMR assistance before in all my carrier. I definitively give to iNMR my strongest recommendation.
Monday, 8 June 2009
Arthur Roberts
Q. Please introduce yourself to the readers of the NMR software blog.
A. I am a project scientist at the School of Pharmacy at the University of California San Diego (UCSD). I was hired to bring some novel NMR technology that I developed at the University of Washington to UCSD.
I worked as a postdoctoral fellow at the Department of Medicinal Chemistry at the University of Washington and at Washington State University. I started my career as an EPR spectroscopist, where we built instruments. I have been doing NMR, since 2003.
Currently I study the process of drug metabolism, which happens to be the main road block for drug development. We hope that our research will lead to drugs of higher efficacy and fewer side effects. We are developing NMR technology that will allow us to rapidly determine drug bound structures and will speed drug development. We have developed a variety of NMR pulse programs for this purpose.
We do Paramagnetic protein NMR. We also do NMR simulations and write NMR pulse programs.
Q. Which NMR software are you using now?
A. Topspin 2.1 and iNMR 3.15.
Q. Which other NMR software have you used in the past?
A. VNMRJ, Spinworks, Sparky, NMRpipe, MestreC, xwinnmr, and MestreNova
Q. How do you rate iNMR?
A. In terms of NMR software, it is the best in terms of ease of use and power.
iNMR
It can process 1D, 2D, and 3D. Easy to use and powerful. It can read multiple formats and can convert files to ascii. The graphics are also very nice. No apparent bugs.
Topspin
It can process 1D, 2D, and 3D. Also powerful, but not very easy to use. I need my data converted to ascii for analysis with other programs and I could not find a way to do it with this software. It can only read Bruker formats.
Mestre-C
It can process 1D and 2D data. Powerful and easy to use, but a little buggy. For 2D, the conversion to ascii is not ideal.
NMRpipe
It can process 1D and 2D data. Not as powerful as the above programs and very clumsy to use. No easy way to convert data to ascii. It is also very slow.
MestreNova
It can process at least 1D and 2D. Powerful and easy to use, but slow, very slow. I found no easy way to convert my 2D spectra to ascii. Also, several useful features were removed from MestreC for this version.
VNMRJ
It can process 1D and 2D. Not as good as Topspin, but equally difficult. This software can not convert to ascii or read other file formats.
Spinworks
It can process 1D and 2D. It is fairly easy to use, but not as powerful as the software above. It is also quite buggy.
This is how I rate all the software and I tested a lot of NMR software:
iNMR > Topspin > Mestre-C > xwinnmr > MestreNova > Spinworks > Sparky > NMRpipe
Q. Is iNMR enough for your needs?
A. Yes, it does everything that I need including processing 3D data sets and it does it fast. It allows me to read files that I produced at the University of Washington on a Varian Unity Inova and the Bruker Avance III at the University of California San Diego. It allows me to convert files to ascii, so that I can do singular value decomposition of it with a scientific analysis program that we use. It produces publication-quality graphics. It is also very easy to use, so I don't need to spend a lot of time training graduate students or other postdocs on how to use it. I also didn't have to spend a lot of time learning it myself. I can't imagine a lab without it.
A. I am a project scientist at the School of Pharmacy at the University of California San Diego (UCSD). I was hired to bring some novel NMR technology that I developed at the University of Washington to UCSD.
I worked as a postdoctoral fellow at the Department of Medicinal Chemistry at the University of Washington and at Washington State University. I started my career as an EPR spectroscopist, where we built instruments. I have been doing NMR, since 2003.
Currently I study the process of drug metabolism, which happens to be the main road block for drug development. We hope that our research will lead to drugs of higher efficacy and fewer side effects. We are developing NMR technology that will allow us to rapidly determine drug bound structures and will speed drug development. We have developed a variety of NMR pulse programs for this purpose.
We do Paramagnetic protein NMR. We also do NMR simulations and write NMR pulse programs.
Q. Which NMR software are you using now?
A. Topspin 2.1 and iNMR 3.15.
Q. Which other NMR software have you used in the past?
A. VNMRJ, Spinworks, Sparky, NMRpipe, MestreC, xwinnmr, and MestreNova
Q. How do you rate iNMR?
A. In terms of NMR software, it is the best in terms of ease of use and power.
iNMR
It can process 1D, 2D, and 3D. Easy to use and powerful. It can read multiple formats and can convert files to ascii. The graphics are also very nice. No apparent bugs.
Topspin
It can process 1D, 2D, and 3D. Also powerful, but not very easy to use. I need my data converted to ascii for analysis with other programs and I could not find a way to do it with this software. It can only read Bruker formats.
Mestre-C
It can process 1D and 2D data. Powerful and easy to use, but a little buggy. For 2D, the conversion to ascii is not ideal.
NMRpipe
It can process 1D and 2D data. Not as powerful as the above programs and very clumsy to use. No easy way to convert data to ascii. It is also very slow.
MestreNova
It can process at least 1D and 2D. Powerful and easy to use, but slow, very slow. I found no easy way to convert my 2D spectra to ascii. Also, several useful features were removed from MestreC for this version.
VNMRJ
It can process 1D and 2D. Not as good as Topspin, but equally difficult. This software can not convert to ascii or read other file formats.
Spinworks
It can process 1D and 2D. It is fairly easy to use, but not as powerful as the software above. It is also quite buggy.
This is how I rate all the software and I tested a lot of NMR software:
iNMR > Topspin > Mestre-C > xwinnmr > MestreNova > Spinworks > Sparky > NMRpipe
Q. Is iNMR enough for your needs?
A. Yes, it does everything that I need including processing 3D data sets and it does it fast. It allows me to read files that I produced at the University of Washington on a Varian Unity Inova and the Bruker Avance III at the University of California San Diego. It allows me to convert files to ascii, so that I can do singular value decomposition of it with a scientific analysis program that we use. It produces publication-quality graphics. It is also very easy to use, so I don't need to spend a lot of time training graduate students or other postdocs on how to use it. I also didn't have to spend a lot of time learning it myself. I can't imagine a lab without it.
Stefano Antoniutti
Q. Please introduce yourself to the readers of the NMR software blog.
A. My position is Associate Professor in General and Inorganic Chemistry since year 2000 at the Dept. of Chemistry, Università di Venezia Ca' Foscari, Italy. I am responsible for Dept. NMR instrumentation and services since Year 1992. I entered the Department in 1983 as a researcher. Since 1983 I have my name in more than 80 scientific papers on ISI classified journals in the field.
My research area is in the field of Inorganic and Organometallic transition metal complexes. The research group I belong to is involved mainly in synthesis and characterization of new complexes.
NMR is the main way of characterization of our new compounds. I began using permanent magnet-pen plotter-1H CW instruments in the early '80s; moved to monodimensional FT NMR in the '80s; today use of multidimensional, multinuclear NMR (1H, 31P, 13C, 15N, 119Sn etc) is my daily routine.
Since 1995 in our Dept. we separated acquisition of spectra from spectral data elaboration, exporting NMR spectra from our Bruker AC instrument to our personal computers, mainly Macintosh machines.
Luckily, after the demise of Bruker from the Mac software area, we discovered the SwaN-MR package (free !!!) which became quickly the workhorse of our daily NMR duties. Today, our Mac users have switched to iNMR, the Wintel users to MNova.
Q. Which other NMR software have you used?
A. Apart from the built-in software of our instruments (Varian and Bruker NMRs) we began in the '90 with WINNMR, a Bruker software written for both PCs and Macs, in two versions. Some day, Buker decided not to develop any more the Mac version, concentrating their efforts only on the PC side. More or less in the same period I discovered SwaN-MR and Giuseppe Balacco, starting a brand new era for our work: you should remember that Bruker software was very expensive (any license was about €500, in those years, and used a hardware key!), and sadly far from complete, at least initially. For example, only 1H and 13C nuclei were supposed to be used; only after some request to the developers' team it was possible to obtain an improved version, really multinuclear!
In a sense, SwaN-MR (which I still use sometime today on a G5 machine) was a complete breakthrough, having a revolutionary impact on our work!
Giuseppe was very cooperative, so everytime some bug was evident, I obtained in a short time (from hours to minutes !) an improved and corrected version. He introduced the simulation routine on my request, and tailored it exactly as a chemist, in my opinion, needed it, not like a software engineer thinks a chemist should use it: a real dream. The same approach he maintained, and improved, when he wrote iNMR.
I find the latter a very high quality software, almost unbeatable for his price/performance ratio. Surely it can be that a more complete software package exists, but at least at ten or more times the price! (If I remember, current Bruker PC software is in the Thousands € range for a license, and only or PC or Linux boxes).
Q. Is iNMR enough for your needs?
A. As usual, you use only a small fraction of the opportunities offered by a software; even after having been host of Giuseppe for a couple of times in the last years, to make him to teach us how to use the program, I think that an usage of more than 20% of the opportunity it offers (and are continuously improved) is to be considered unrealistc. Everyday I discover something new, and with more or less the same frequency, new options are offered by new versions of the software, which Giuseppe considers really a commitment to do.
Anyway, iNMR never failed to offer me a solution to any need in my research work, and I always cite it in my papers, hoping to extend the number of its user. I evaluated other software, but none fulfilled my needs like iNMR (and, in the times of Mac OS 7/8/9, SwaN-MR). For instance, its baseline correction routine in bidimensional spectra is outstanding, far better in results than the Winnmr one, letting you extract correlations you would have missed otherwise.
A. My position is Associate Professor in General and Inorganic Chemistry since year 2000 at the Dept. of Chemistry, Università di Venezia Ca' Foscari, Italy. I am responsible for Dept. NMR instrumentation and services since Year 1992. I entered the Department in 1983 as a researcher. Since 1983 I have my name in more than 80 scientific papers on ISI classified journals in the field.
My research area is in the field of Inorganic and Organometallic transition metal complexes. The research group I belong to is involved mainly in synthesis and characterization of new complexes.
NMR is the main way of characterization of our new compounds. I began using permanent magnet-pen plotter-1H CW instruments in the early '80s; moved to monodimensional FT NMR in the '80s; today use of multidimensional, multinuclear NMR (1H, 31P, 13C, 15N, 119Sn etc) is my daily routine.
Since 1995 in our Dept. we separated acquisition of spectra from spectral data elaboration, exporting NMR spectra from our Bruker AC instrument to our personal computers, mainly Macintosh machines.
Luckily, after the demise of Bruker from the Mac software area, we discovered the SwaN-MR package (free !!!) which became quickly the workhorse of our daily NMR duties. Today, our Mac users have switched to iNMR, the Wintel users to MNova.
Q. Which other NMR software have you used?
A. Apart from the built-in software of our instruments (Varian and Bruker NMRs) we began in the '90 with WINNMR, a Bruker software written for both PCs and Macs, in two versions. Some day, Buker decided not to develop any more the Mac version, concentrating their efforts only on the PC side. More or less in the same period I discovered SwaN-MR and Giuseppe Balacco, starting a brand new era for our work: you should remember that Bruker software was very expensive (any license was about €500, in those years, and used a hardware key!), and sadly far from complete, at least initially. For example, only 1H and 13C nuclei were supposed to be used; only after some request to the developers' team it was possible to obtain an improved version, really multinuclear!
In a sense, SwaN-MR (which I still use sometime today on a G5 machine) was a complete breakthrough, having a revolutionary impact on our work!
Giuseppe was very cooperative, so everytime some bug was evident, I obtained in a short time (from hours to minutes !) an improved and corrected version. He introduced the simulation routine on my request, and tailored it exactly as a chemist, in my opinion, needed it, not like a software engineer thinks a chemist should use it: a real dream. The same approach he maintained, and improved, when he wrote iNMR.
I find the latter a very high quality software, almost unbeatable for his price/performance ratio. Surely it can be that a more complete software package exists, but at least at ten or more times the price! (If I remember, current Bruker PC software is in the Thousands € range for a license, and only or PC or Linux boxes).
Q. Is iNMR enough for your needs?
A. As usual, you use only a small fraction of the opportunities offered by a software; even after having been host of Giuseppe for a couple of times in the last years, to make him to teach us how to use the program, I think that an usage of more than 20% of the opportunity it offers (and are continuously improved) is to be considered unrealistc. Everyday I discover something new, and with more or less the same frequency, new options are offered by new versions of the software, which Giuseppe considers really a commitment to do.
Anyway, iNMR never failed to offer me a solution to any need in my research work, and I always cite it in my papers, hoping to extend the number of its user. I evaluated other software, but none fulfilled my needs like iNMR (and, in the times of Mac OS 7/8/9, SwaN-MR). For instance, its baseline correction routine in bidimensional spectra is outstanding, far better in results than the Winnmr one, letting you extract correlations you would have missed otherwise.
Sunday, 31 May 2009
Testing the Razors
As I wrote initially, I like the razors because they are easy to learn and use.
I have simulated two molecules only, because they are the standard (minimal) test I perform on simulation software. I come from the old school, where simulation means generating a plot from chemical shift values (while the razors mainly estimate the chemical shifts from the structure).
The first test is N,N-dimethylformamide.
The second test is ortho-dichloro-benzene.
Simulated by iNMR:
Simulated by HNMRazor:
I have simulated two molecules only, because they are the standard (minimal) test I perform on simulation software. I come from the old school, where simulation means generating a plot from chemical shift values (while the razors mainly estimate the chemical shifts from the structure).
The first test is N,N-dimethylformamide.
1-H
exp. calc. diff.
8.019 1.609 -6.41
2.970 3.001 0.031
2.883 3.001 0.118
13-C
exp. calc. diff.
162.6 198.7 36.1
36.4 34.568 -1.9
31.3 34.568 3.3
The second test is ortho-dichloro-benzene.
Simulated by iNMR:

Simulated by HNMRazor:

Interview with Kevin Theisen
Kevin already told his story on the offcial website of iChemLabs. I was curious to know more details... Here is the first offical interview of my blog.
OS: How much accurate are the predictions of the NMRazors?
KT: The NMRazors are fairly accurate for most molecules. They will handle any molecule encountered in an undergraduate organic chemistry course. The NMRazors will be less accurate for molecules where complex anisotropic and 3D effects are present. I used several published references when developing the algorithms in the NMRazors and they are cited on the NMRazor website.
OS: Programming is similar to chemical synthesis: there are starting materials and finished products. What were the staring material for the NMRazors?
KT: When I first began programming chemistry applications, I started with a graph based depth-first search traversal of a database of reactions in order to optimize synthetic routes. A credible synthetic database was too expensive for me to obtain as an undergraduate, so I moved on to other applications in chemistry. I quickly discovered that the graph data structure is really integral to computational chemistry, as most chemical entities are efficiently modeled with them, especially structures. I was and still am a huge fan of spectroscopy, so I began to work on algorithms to traverse molecules and find functional groups for nuclear magnetic resonance simulations. It was originally a text based application, and I remember showing some of my favorite professors connection table inputs with ppm table outputs. It was very unattractive, so I taught myself Swing and the NMRazor GUI was created.
OS: Why do you prefer Java? Because of the language itself, the available frameworks, the platform independence or any other reason?
KT: Java is a wonderful programming language for several reasons. Mainly, it's object oriented and the graphical capabilities available with Java Swing are really unparalleled in other languages. The other reason was that I used a Mac, my friends usually used PCs and a few had Linux, so I needed a programming language that I could use on Mac and then deploy on other operating systems. Java was really the only choice for me at the time, given my minimal experience. The only downside to Java is that it is interpreted, so it may be slower if the program is carelessly written, and the JREs on different operating systems are not always consistent, so I still need to test on all three systems before I am sure a program actually works.
OS: How much work was required? What was the most difficult part: the algorithm or the interface?
KT: The interface was the most difficult part of the NMRazors because I was just starting to learn Java Swing. Now that I have pretty much mastered Java Swing, perfecting the algorithms is more difficult and takes far more time. But this is a good thing, because this saved time I can devote to truly perfecting the algorithms.
OS: Can you briefly describe the algorithms?
KT: There are two main ways to predict spectra, quantum mechanics and chemoinformatics. Quantum mechanics approaches calculate electron densities and then predict chemical shifts based on shielding. The NMRazors use a chemoinformatics approach, which takes a large database of spectra, defines similarity between the structures, and then interpolates to predict the chemical shifts of unknown species. Both can be very accurate if done properly, but quantum approaches take several hours. Since I couldn't afford a database of spectra, I went one level of abstraction further and used incremental constants to predict shifts. So the algorithm is similar to how a spectroscopist would predict a chemical shift in their head: they would look at the functional groups surrounding the nucleus of interest and add together the deshielding effects. There are other approximations for solvent effects, and splitting needs to be calculated, as well as second order interactions and more. These considerations make sure the simulated spectrum is as close to the real spectrum as possible.
OS: How do you manage to study and lead iChemlabs at the same time?
KT: There are days where it gets a little difficult. But fortunately, I have help with all the backend servers and services, web design, software testing, and of course accountants to do all the financials. Also, we were careful when we set up the company to put the appropriate infrastructure in place to manage our growth. Because we made sure to have redundant servers and source code control and bug reporting systems and product build processes in place, adding new products has become routine. Furthermore we outsource email and payment services so we just don't have to deal with those headaches. The bottom line, though, is I am very passionate about my company and my research. Currently, my research has become very exciting. It is a mix of chemical informatics and theory, which provide daily puzzles for me to solve. Both iChemLabs and my research push me to different edges of technology. Since I really enjoy both jobs it is not that hard to keep them both going. Every day I simply have fun with this stuff.
OS: You are marketing the razors for educational use. Could there be any practical use for them?
KT: My goal in my work is to provide quality software to students and scientists that is both accessible and affordable. The NMRazors were released for free for educational use because I knew it would be useful for students like myself that were working hard trying to interpret spectra for the first time. I also price our other software affordably so that I can continue to fund their development, without requiring customers to spend half of their savings. The NMRazors are used at several universities around the world now, and I am very happy with how this project developed.
In terms of practicality, as the algorithms mature over time, they will certainly be very useful to industry.
OS: Let's talk about ChemDoodle. How do you compare it against IsisDraw, ACD ChemSketch and Marvin?
KT: Firstly, I created ChemDoodle because I needed a very robust set of features, I could not afford ChemDraw, and ACD Labs would not respond to my emails when I requested pricing information.
Regardless, the chemical drawing software that was available was built on 10-20 year old systems that are severely out of date. ChemDoodle takes a new approach to drawing molecules, making the interface aesthetic and the controls more intuitive, providing functionality that takes advantage of current technology such as connections to online databases, and providing a more artistic approach to drawing figures by allowing users to completely control the look of structures including using different bond stroke styles for truly stunning graphics. ChemDoodle also contains many widgets, or mini-applications, which are added bonuses that perform very powerful and specific tasks. All these reasons are why I refer to ChemDoodle as a chemical structure environment, rather than just a chemical drawing tool like the ones mentioned.
Two years later, and we have succeeded in solving many of the issues other programs suffer from, and we work wonderfully on all operating systems. We receive emails almost daily complimenting us on our work and for creating a superior chemical drawing program, and we proudly agree.
OS: Are you going to write software for the iPhone? It seems like half of the world is doing it...
KT: That was a very attractive idea a year ago, when Brad Larson created Molecules. However, we are really focused on perfecting our desktop software and pushing ChemDoodle to be the best chemical drawing program in everyone's mind. We also have a couple secret projects that are under development, though not for the iPhone. These are brand new ideas, very different from current software, and we are sure chemists will love to use them.
OS: How much accurate are the predictions of the NMRazors?
KT: The NMRazors are fairly accurate for most molecules. They will handle any molecule encountered in an undergraduate organic chemistry course. The NMRazors will be less accurate for molecules where complex anisotropic and 3D effects are present. I used several published references when developing the algorithms in the NMRazors and they are cited on the NMRazor website.
OS: Programming is similar to chemical synthesis: there are starting materials and finished products. What were the staring material for the NMRazors?
KT: When I first began programming chemistry applications, I started with a graph based depth-first search traversal of a database of reactions in order to optimize synthetic routes. A credible synthetic database was too expensive for me to obtain as an undergraduate, so I moved on to other applications in chemistry. I quickly discovered that the graph data structure is really integral to computational chemistry, as most chemical entities are efficiently modeled with them, especially structures. I was and still am a huge fan of spectroscopy, so I began to work on algorithms to traverse molecules and find functional groups for nuclear magnetic resonance simulations. It was originally a text based application, and I remember showing some of my favorite professors connection table inputs with ppm table outputs. It was very unattractive, so I taught myself Swing and the NMRazor GUI was created.
OS: Why do you prefer Java? Because of the language itself, the available frameworks, the platform independence or any other reason?
KT: Java is a wonderful programming language for several reasons. Mainly, it's object oriented and the graphical capabilities available with Java Swing are really unparalleled in other languages. The other reason was that I used a Mac, my friends usually used PCs and a few had Linux, so I needed a programming language that I could use on Mac and then deploy on other operating systems. Java was really the only choice for me at the time, given my minimal experience. The only downside to Java is that it is interpreted, so it may be slower if the program is carelessly written, and the JREs on different operating systems are not always consistent, so I still need to test on all three systems before I am sure a program actually works.
OS: How much work was required? What was the most difficult part: the algorithm or the interface?
KT: The interface was the most difficult part of the NMRazors because I was just starting to learn Java Swing. Now that I have pretty much mastered Java Swing, perfecting the algorithms is more difficult and takes far more time. But this is a good thing, because this saved time I can devote to truly perfecting the algorithms.
OS: Can you briefly describe the algorithms?
KT: There are two main ways to predict spectra, quantum mechanics and chemoinformatics. Quantum mechanics approaches calculate electron densities and then predict chemical shifts based on shielding. The NMRazors use a chemoinformatics approach, which takes a large database of spectra, defines similarity between the structures, and then interpolates to predict the chemical shifts of unknown species. Both can be very accurate if done properly, but quantum approaches take several hours. Since I couldn't afford a database of spectra, I went one level of abstraction further and used incremental constants to predict shifts. So the algorithm is similar to how a spectroscopist would predict a chemical shift in their head: they would look at the functional groups surrounding the nucleus of interest and add together the deshielding effects. There are other approximations for solvent effects, and splitting needs to be calculated, as well as second order interactions and more. These considerations make sure the simulated spectrum is as close to the real spectrum as possible.
OS: How do you manage to study and lead iChemlabs at the same time?
KT: There are days where it gets a little difficult. But fortunately, I have help with all the backend servers and services, web design, software testing, and of course accountants to do all the financials. Also, we were careful when we set up the company to put the appropriate infrastructure in place to manage our growth. Because we made sure to have redundant servers and source code control and bug reporting systems and product build processes in place, adding new products has become routine. Furthermore we outsource email and payment services so we just don't have to deal with those headaches. The bottom line, though, is I am very passionate about my company and my research. Currently, my research has become very exciting. It is a mix of chemical informatics and theory, which provide daily puzzles for me to solve. Both iChemLabs and my research push me to different edges of technology. Since I really enjoy both jobs it is not that hard to keep them both going. Every day I simply have fun with this stuff.
OS: You are marketing the razors for educational use. Could there be any practical use for them?
KT: My goal in my work is to provide quality software to students and scientists that is both accessible and affordable. The NMRazors were released for free for educational use because I knew it would be useful for students like myself that were working hard trying to interpret spectra for the first time. I also price our other software affordably so that I can continue to fund their development, without requiring customers to spend half of their savings. The NMRazors are used at several universities around the world now, and I am very happy with how this project developed.
In terms of practicality, as the algorithms mature over time, they will certainly be very useful to industry.
OS: Let's talk about ChemDoodle. How do you compare it against IsisDraw, ACD ChemSketch and Marvin?
KT: Firstly, I created ChemDoodle because I needed a very robust set of features, I could not afford ChemDraw, and ACD Labs would not respond to my emails when I requested pricing information.
Regardless, the chemical drawing software that was available was built on 10-20 year old systems that are severely out of date. ChemDoodle takes a new approach to drawing molecules, making the interface aesthetic and the controls more intuitive, providing functionality that takes advantage of current technology such as connections to online databases, and providing a more artistic approach to drawing figures by allowing users to completely control the look of structures including using different bond stroke styles for truly stunning graphics. ChemDoodle also contains many widgets, or mini-applications, which are added bonuses that perform very powerful and specific tasks. All these reasons are why I refer to ChemDoodle as a chemical structure environment, rather than just a chemical drawing tool like the ones mentioned.
Two years later, and we have succeeded in solving many of the issues other programs suffer from, and we work wonderfully on all operating systems. We receive emails almost daily complimenting us on our work and for creating a superior chemical drawing program, and we proudly agree.
OS: Are you going to write software for the iPhone? It seems like half of the world is doing it...
KT: That was a very attractive idea a year ago, when Brad Larson created Molecules. However, we are really focused on perfecting our desktop software and pushing ChemDoodle to be the best chemical drawing program in everyone's mind. We also have a couple secret projects that are under development, though not for the iPhone. These are brand new ideas, very different from current software, and we are sure chemists will love to use them.
Sunday, 24 May 2009
Unusual Solution
This is how I solved the problem of the razor.
1- I described the bug on my (this) blog.
2- It rang a bell into Kevin's mind;
3- Kevin recompiled both programs (1-H and 13-C).
It may sound difficult for you, yet recompiling a program is very easy (provided you are the author, it can be a nightmare in other cases!).
The new versions work and produce the desired plots.
I will return on the subject with more articles.
Thank you, Kevin!
1- I described the bug on my (this) blog.
2- It rang a bell into Kevin's mind;
3- Kevin recompiled both programs (1-H and 13-C).
It may sound difficult for you, yet recompiling a program is very easy (provided you are the author, it can be a nightmare in other cases!).
The new versions work and produce the desired plots.
I will return on the subject with more articles.
Thank you, Kevin!
We Insist!
Maybe there is an error into the drawing module, or I haven't understood how to use it.
Today I am going to bypass drawing with a clever trick. You can follow me step by step.
1) I open NHMRazor.
2) Command: Find Molecule.
3) I insert a molecular formula: C6H12.
4) I find 30 result: great! I scroll down the list and select 2-methyl-2-ene.
5) I click the button "Load". This loads the structure:
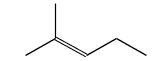 We have almost done it! Next step:
We have almost done it! Next step:
6) A click on the triplet icon.
7) This time I don't select any solvent (who knows?) and assume that the default settings are OK. Final step: button OK...
I am not showing the result because nothing has changed since yesterday ("The server has an error that is currently being fixed. Sorry for any inconvenience").
Have you ever played Myst? You know the feeling.
Today I am going to bypass drawing with a clever trick. You can follow me step by step.
1) I open NHMRazor.
2) Command: Find Molecule.
3) I insert a molecular formula: C6H12.
4) I find 30 result: great! I scroll down the list and select 2-methyl-2-ene.
5) I click the button "Load". This loads the structure:
 We have almost done it! Next step:
We have almost done it! Next step:6) A click on the triplet icon.
7) This time I don't select any solvent (who knows?) and assume that the default settings are OK. Final step: button OK...
I am not showing the result because nothing has changed since yesterday ("The server has an error that is currently being fixed. Sorry for any inconvenience").
Have you ever played Myst? You know the feeling.
Saturday, 23 May 2009

Shaving with a new HNMRazor
The NMR razors are simple and (probably) powerful applications that can simulate a spectrum directly from the molecular formula. For the user's point of view it works like a (very) simplified version of ChemDraw. When you have finished drawing the structure, you hit a button to simulate the spectrum. When you hit the button, the program asks you such spectroscopic details like solvent, temperature, magnetic field, etc.. When you have finished this further step, the razor calls home. Yes, because all you have downloaded is a graphic interface, the true program is running into a remote and ultra-secret location.
I like this program because I have understood everything in 5 minutes.
To start with, I have simulated the 1-H spectrum of ethyl acetate in CDCl3, at 200 MHz.
Here it is:
 Well, you know, I am not a lucky guy! Enough for today. If you want to try by yourself:
Well, you know, I am not a lucky guy! Enough for today. If you want to try by yourself:
http://www.ichemlabs.com/content/nmrazors
This morning I shaved my face with a Gillette blue II. It is as good as a Gillette can be and it is much cheaper than the other models of the same brand. Officially it is disposable, yet I don't remember when I bought it. Quite likely I have been using the same razor for 3 or 4 months. As long as it works... Two complete reviews in a single post, I am very productive...
I like this program because I have understood everything in 5 minutes.
To start with, I have simulated the 1-H spectrum of ethyl acetate in CDCl3, at 200 MHz.
Here it is:
 Well, you know, I am not a lucky guy! Enough for today. If you want to try by yourself:
Well, you know, I am not a lucky guy! Enough for today. If you want to try by yourself:http://www.ichemlabs.com/content/nmrazors
This morning I shaved my face with a Gillette blue II. It is as good as a Gillette can be and it is much cheaper than the other models of the same brand. Officially it is disposable, yet I don't remember when I bought it. Quite likely I have been using the same razor for 3 or 4 months. As long as it works... Two complete reviews in a single post, I am very productive...
Friday, 8 May 2009

Monday, 20 April 2009
Tuesday, 7 April 2009
one look
My software always shows me the FID when I open a new spectrum. Then I transform it. Some people find this ritual annoying and prefer to see the transformed spectrum directly, because all the FIDs look the same. Sometimes it happens, however, that a FID looks different:
 It merely means that something wrong happened during the acquisition. No matter what went wrong, no matter why this particular FID looks different. It is enough to recognize that it is very unusual.
It merely means that something wrong happened during the acquisition. No matter what went wrong, no matter why this particular FID looks different. It is enough to recognize that it is very unusual.
Having a look at the FID is not a waste of time. Processing this particular FID and trying in all ways to compensate for its defects can potentially be a waste of time.
My software always shows me the FID because I want it so.
 It merely means that something wrong happened during the acquisition. No matter what went wrong, no matter why this particular FID looks different. It is enough to recognize that it is very unusual.
It merely means that something wrong happened during the acquisition. No matter what went wrong, no matter why this particular FID looks different. It is enough to recognize that it is very unusual.Having a look at the FID is not a waste of time. Processing this particular FID and trying in all ways to compensate for its defects can potentially be a waste of time.
My software always shows me the FID because I want it so.
Wednesday, 1 April 2009
Sweet J
It took 14 years for Sweet J to grow.
When only a few hard-die fans could remember the name, version 2.0 appeared. The new interface resembles the old one, yet the differences are noteworthy. Version 1 included 4 modal dialogs. Now only the preferences dialog has survived. The other three have been fused, instead, into the main window. The interface updates immediately, therefore there is no "OK" button. The purpose of Sweet J is limited. The implementation, instead, is rather sophisticated. I say it because the source code is freely available.
When I say "sophisticated" I mean that it's not trivial to connect all the pieces of the interface to a chemistry logic. For example, if you change one of the skeleton atoms, the equation by Haasnoot et al. is no more applicable, therefore the whole interface needs to be updated (some controls disappear, for example).
The picture below has already appeared on the Apple site, on Macupdate and on Macs in Chemistry.
 My blog is the official sponsor of this effort. Or is it the opposite? (Sweet J sponsoring the NMR software blog?)... There's also a theory going around which says that Lou Reed wrote a song inspired by it, which is unbelievable: "Sweet Jane" the song predates "Sweet J" the program. Maybe it's the opposite...
My blog is the official sponsor of this effort. Or is it the opposite? (Sweet J sponsoring the NMR software blog?)... There's also a theory going around which says that Lou Reed wrote a song inspired by it, which is unbelievable: "Sweet Jane" the song predates "Sweet J" the program. Maybe it's the opposite...
When only a few hard-die fans could remember the name, version 2.0 appeared. The new interface resembles the old one, yet the differences are noteworthy. Version 1 included 4 modal dialogs. Now only the preferences dialog has survived. The other three have been fused, instead, into the main window. The interface updates immediately, therefore there is no "OK" button. The purpose of Sweet J is limited. The implementation, instead, is rather sophisticated. I say it because the source code is freely available.
When I say "sophisticated" I mean that it's not trivial to connect all the pieces of the interface to a chemistry logic. For example, if you change one of the skeleton atoms, the equation by Haasnoot et al. is no more applicable, therefore the whole interface needs to be updated (some controls disappear, for example).
The picture below has already appeared on the Apple site, on Macupdate and on Macs in Chemistry.
 My blog is the official sponsor of this effort. Or is it the opposite? (Sweet J sponsoring the NMR software blog?)... There's also a theory going around which says that Lou Reed wrote a song inspired by it, which is unbelievable: "Sweet Jane" the song predates "Sweet J" the program. Maybe it's the opposite...
My blog is the official sponsor of this effort. Or is it the opposite? (Sweet J sponsoring the NMR software blog?)... There's also a theory going around which says that Lou Reed wrote a song inspired by it, which is unbelievable: "Sweet Jane" the song predates "Sweet J" the program. Maybe it's the opposite...
Sunday, 22 March 2009
Prima Cala

This beach is near to my home (a 5 minutes walk). I come here whenever I need a little of fresh hair. The place is often windy, like today.
Friday, 20 March 2009

Thursday, 19 March 2009

Friday, 13 March 2009
SDBS beautified
The Spectral Database for Organic Compounds is very popular
and deserves all my praise: is straightforward to use, easy to access, rich of data, void of advertisement and completely free. Everybody has used it at least once, what else should I say?
Jaume Farràs Soler sent me a shell script to import a list of peaks, copied from SDBS, into iNMR. He said that the pictures of SDBS were not clear enough, while in this way he could generate much better ones. The address of this script is now:
http://www.inmr.net/library/sdbs2inmr
Later on we'll see why it can be useful. Let's start with a more immediate approach. First thing first: if you have a Mac, download the latest version (3.1.4) of iNMR passion (freeware). Now, the next time you find a 1-H spectrum on SDBS, scroll down the page, until you find this button: Clicking the button leads you to a list like:
Clicking the button leads you to a list like:
Copy the list, including the header: "Hz ppm Int.", into the clipboard. Open iNMR passion and choose, from the menu, "Simulate List of Peaks". You will instantly see the spectrum, synthesized, as large as your monitor, ready to be explored (or printed) and, it goes without saying, anti-aliased.
Caveat: I guess that SDBS reports the apparent eight of the peaks, which is given by the proper height of a peak plus the contribute of any overlapping peaks. iNMR interprets this value, instead, as the natural height. The result is that all the peaks of a multiplet will become taller, while the singlets will be OK.
The areas will be a little distorted. What's also missing from the SDBS list is the width of each peak. In the synthetic spectrum all lines appear equal and narrow (1 Hz wide). The areas of broad peaks will be strongly underestimated. Add to this the effect above (for overlapping peaks).
If you want something more realistic, you can use the script by Jaume. It will generate a list with widths. Import the list into the deconvolution module of iNMR, where you'll be able to modify individual line-widths and intensities. The spectrum will look more realistic, yet don't ask me where to get the correct values from.
If, instead, you know about another database, with a different list format, send me the address. I'll write another version of iNMR passion that will recognize your format too.
and deserves all my praise: is straightforward to use, easy to access, rich of data, void of advertisement and completely free. Everybody has used it at least once, what else should I say?
Jaume Farràs Soler sent me a shell script to import a list of peaks, copied from SDBS, into iNMR. He said that the pictures of SDBS were not clear enough, while in this way he could generate much better ones. The address of this script is now:
http://www.inmr.net/library/sdbs2inmr
Later on we'll see why it can be useful. Let's start with a more immediate approach. First thing first: if you have a Mac, download the latest version (3.1.4) of iNMR passion (freeware). Now, the next time you find a 1-H spectrum on SDBS, scroll down the page, until you find this button:
 Clicking the button leads you to a list like:
Clicking the button leads you to a list like:
Hz ppm Int.
1367.55 3.422 485
1360.72 3.405 942
1353.88 3.388 515
755.00 1.890 118
.......................Copy the list, including the header: "Hz ppm Int.", into the clipboard. Open iNMR passion and choose, from the menu, "Simulate List of Peaks". You will instantly see the spectrum, synthesized, as large as your monitor, ready to be explored (or printed) and, it goes without saying, anti-aliased.
Caveat: I guess that SDBS reports the apparent eight of the peaks, which is given by the proper height of a peak plus the contribute of any overlapping peaks. iNMR interprets this value, instead, as the natural height. The result is that all the peaks of a multiplet will become taller, while the singlets will be OK.
The areas will be a little distorted. What's also missing from the SDBS list is the width of each peak. In the synthetic spectrum all lines appear equal and narrow (1 Hz wide). The areas of broad peaks will be strongly underestimated. Add to this the effect above (for overlapping peaks).
If you want something more realistic, you can use the script by Jaume. It will generate a list with widths. Import the list into the deconvolution module of iNMR, where you'll be able to modify individual line-widths and intensities. The spectrum will look more realistic, yet don't ask me where to get the correct values from.
If, instead, you know about another database, with a different list format, send me the address. I'll write another version of iNMR passion that will recognize your format too.
Tuesday, 10 March 2009
Evgeny wants you
Last year he opened the NMR wiki. This month Evgeny Fadeev has started a discussion group. Synthetic description: "All about magnetic resonance, summaries go to NMR Wiki". Evgeny also says: "Send invitations to your friends and ask them to invite their friends. Then we'll make it work!!!".
You are in time to become one of the first 100 members.

You are in time to become one of the first 100 members.
Sunday, 8 March 2009
Making the Headlines
The NMR discovery of the month, according to spectroscopynow, is the whitening method. When this humble blog introduced the method in October, I didn't believe it was SO important. I am still pinching myself.

Friday, 6 March 2009
ini386
The reader ini386 compared the ease of use of iNMR with that of the Adobe reader. Such a comparison is impossible. Both products and their histories are under the eyes of every one. You should know them! Today I am trying to expose a few facts that are never been secret, they have been just ignored. In other words: nobody really cares about what I am going to write.
iNMR has arrived to version 3.1.1, with 3.1.3 ready to appear next week, through an incredible number of changes and upgrades. In the beginning, the process could be explained in two ways: either the preliminary versions were not satisfactory or the users were impossible to satisfy (does it make any difference?). Afterwards the process became a little different. Once you know that it's so easy to release a new version, it's impossible to stop. You are happy with the program not because it's perfect, but because you know that any defect you find can be eliminated.
While there is a single author, iNMR is the product of many minds. Two hands only; many minds. The clear separation of roles is a winning strategy. The users are in command: their desiderata are normally satisfied. The exception is when they ask another application or a companion application; these requests are difficult to satisfy. If, instead, they ask for a new functionality or a single modification, they are the kings. The user, however, don't the internal mechanism of the program and can't imagine them. It was relatively easy in the case of SwaN-MR, where the data structures were the same (in RAM, on disc and into the dialogs). It's impossible in the case of iNMR: the data structures (which contain the same data) changes at each level and there's a great work of translation under the surface. For this reason the user/king can't tell to the servant how to do his job.
When it's working time, the servant is the king. He can decide, for example, to spend a week to finish a job that could have been done in a couple of hours. The fun is in trying all the possible variations (and not having a deadline). Misunderstandings are frequent (and funny too): the user asks for a certain function, the programmer understands a different thing. Eventually both things are done (and remain; and the program grows...).
The opposite situation (democracy) is dull and stressing. In this case there are the meetings, boring and time-consuming, where there's always people who need to speak even when they have no (useful) idea to communicate. Often you can also encounter their historical enemies, and the psychodrama is on the show. Now, if you know that a new proposal for a change or for an experiment must be approved by a meeting of this kind, you'll be scared of proposing anything (and you'll never desire to experiment).
Don't get me wrong. I know very well that the concentration of resources (energies, capital, skills, everything) is the road to success. I am convinced that I go nowhere if I am alone. What's difficult (and probably never required) is to find an agreement between the coworkers. That's impossible while you are inventing something new. When a problem has already been studied very well, when several books have already been dedicated to the subject, then it becomes possible to work as a team.
Programming is becoming more and more similar to chemistry. When intermediate structures become important and popular, they are prepared commercially. Teams of programmers work together to create general-purpose libraries. Individual programmers can assemble the ready-made pieces to create the final applications. When the closest intermediate can't be found, we can still copy (recycle) the ideas, as we do in chemistry.
I couldn't have written this post 20 years ago. I was a different man. I used to say to the users: "This is the program. Learn it." And they kept saying: "Have you ever though about adding...? Why don't you imitate that other program...?". We have kept saying the same things over and over until today (and tomorrow). What is changed is that now I pay more attention of what they say, while they understand more of what I am saying. In the past I was happy if I could change their minds. Today I am happy when they have changed mine. It's not a virtue, it's my beard that's becoming grey. When I had a whole life in front of me, I tried to make the world a better place where to spend this apparently unlimited existence. I tried to make people think just like me. Today I enjoy to learn new things, to embrace new ideas. It's not as having another chance in life, it doesn't even come close, but it has a flavor of it.
iNMR has arrived to version 3.1.1, with 3.1.3 ready to appear next week, through an incredible number of changes and upgrades. In the beginning, the process could be explained in two ways: either the preliminary versions were not satisfactory or the users were impossible to satisfy (does it make any difference?). Afterwards the process became a little different. Once you know that it's so easy to release a new version, it's impossible to stop. You are happy with the program not because it's perfect, but because you know that any defect you find can be eliminated.
While there is a single author, iNMR is the product of many minds. Two hands only; many minds. The clear separation of roles is a winning strategy. The users are in command: their desiderata are normally satisfied. The exception is when they ask another application or a companion application; these requests are difficult to satisfy. If, instead, they ask for a new functionality or a single modification, they are the kings. The user, however, don't the internal mechanism of the program and can't imagine them. It was relatively easy in the case of SwaN-MR, where the data structures were the same (in RAM, on disc and into the dialogs). It's impossible in the case of iNMR: the data structures (which contain the same data) changes at each level and there's a great work of translation under the surface. For this reason the user/king can't tell to the servant how to do his job.
When it's working time, the servant is the king. He can decide, for example, to spend a week to finish a job that could have been done in a couple of hours. The fun is in trying all the possible variations (and not having a deadline). Misunderstandings are frequent (and funny too): the user asks for a certain function, the programmer understands a different thing. Eventually both things are done (and remain; and the program grows...).
The opposite situation (democracy) is dull and stressing. In this case there are the meetings, boring and time-consuming, where there's always people who need to speak even when they have no (useful) idea to communicate. Often you can also encounter their historical enemies, and the psychodrama is on the show. Now, if you know that a new proposal for a change or for an experiment must be approved by a meeting of this kind, you'll be scared of proposing anything (and you'll never desire to experiment).
Don't get me wrong. I know very well that the concentration of resources (energies, capital, skills, everything) is the road to success. I am convinced that I go nowhere if I am alone. What's difficult (and probably never required) is to find an agreement between the coworkers. That's impossible while you are inventing something new. When a problem has already been studied very well, when several books have already been dedicated to the subject, then it becomes possible to work as a team.
Programming is becoming more and more similar to chemistry. When intermediate structures become important and popular, they are prepared commercially. Teams of programmers work together to create general-purpose libraries. Individual programmers can assemble the ready-made pieces to create the final applications. When the closest intermediate can't be found, we can still copy (recycle) the ideas, as we do in chemistry.
I couldn't have written this post 20 years ago. I was a different man. I used to say to the users: "This is the program. Learn it." And they kept saying: "Have you ever though about adding...? Why don't you imitate that other program...?". We have kept saying the same things over and over until today (and tomorrow). What is changed is that now I pay more attention of what they say, while they understand more of what I am saying. In the past I was happy if I could change their minds. Today I am happy when they have changed mine. It's not a virtue, it's my beard that's becoming grey. When I had a whole life in front of me, I tried to make the world a better place where to spend this apparently unlimited existence. I tried to make people think just like me. Today I enjoy to learn new things, to embrace new ideas. It's not as having another chance in life, it doesn't even come close, but it has a flavor of it.
Thursday, 26 February 2009
Limited Undo
It's great when you can undo after you have already saved a document and go back, back, back... It would be even greater if everything could be really undoable. Take for example Adobe(R) Acrobat(R) Reader 9.0.0. I was reading a book of 1200 pages. I was reading one of the internal pages (don't remember which) and had arrived to the bottom of the window. I moved my hand to press the key "Page Down", but I pressed the wrong (nearby) key, which happens to be the "End" key. Ok, I said, there's the Undo command. No way, said Acrobat Reader, you can't undo. The rationale is probably: "The user has not edited the document, there is nothing to undo". Why not adopting the rationale: "Every time a key is pressed, chances are it was a mistake"???
Dear reader, don't think that, just because today's software allow for unlimited undoing, you can really undo everything. Actually there are a lot of important and common things that can't be undone, or can be remedied to, but only using a command different from Undo. Try for example to click a link in this page and to return here with an Undo. Does it work? Try typing a long sentence with your word processor and to remove the last word only with Undo. What happens?
Luckily, everybody knows how to jump back to the previous page with the command go back and to delete the last word by selecting it and cutting it away. How do you find the page you were reading with Acrobat?
Adobe Reader takes 172.6 MB on my hard disk. It's larger than many historical operative systems (combined). It's also much larger than my first hard disc (40 MB), yet it can't undo. It's really a mystery what's hidden inside. There is potential room for a million of viruses.
I have an assignment for you. I mean you guys who haven't the least idea of what NMR means but nonetheless try to post comments into my blog only to link to your web sites. I have always deleted your posts. This time I want to give you a chance.
Read the credits of Adobe Reader (they appear after you open the "about" box). If you can write the exact number of people who appear into the credits, I will not delete your comment.
Dear reader, don't think that, just because today's software allow for unlimited undoing, you can really undo everything. Actually there are a lot of important and common things that can't be undone, or can be remedied to, but only using a command different from Undo. Try for example to click a link in this page and to return here with an Undo. Does it work? Try typing a long sentence with your word processor and to remove the last word only with Undo. What happens?
Luckily, everybody knows how to jump back to the previous page with the command go back and to delete the last word by selecting it and cutting it away. How do you find the page you were reading with Acrobat?
Adobe Reader takes 172.6 MB on my hard disk. It's larger than many historical operative systems (combined). It's also much larger than my first hard disc (40 MB), yet it can't undo. It's really a mystery what's hidden inside. There is potential room for a million of viruses.
I have an assignment for you. I mean you guys who haven't the least idea of what NMR means but nonetheless try to post comments into my blog only to link to your web sites. I have always deleted your posts. This time I want to give you a chance.
Read the credits of Adobe Reader (they appear after you open the "about" box). If you can write the exact number of people who appear into the credits, I will not delete your comment.
Friday, 6 February 2009
New Millennium
What's the difference between the old and the new millenium? Do you remember when I described the whitening method? Now it has been published, as a peer-reviewd article, on Magnetic Resonance in Chemistry:
Automatic phase correction of 2D NMR spectra by a whitening method
Giuseppe Balacco, Carlos Cobas
Published Online: Feb 3 2009 10:57AM
DOI: 10.1002/mrc.2394
If you have followed this blog, you know that many other papers have appeared on a similar subject (automatic phase correction of 1-D spectra). In the past, to discover that the method didn't actually work, you had to write a program by yourself (apart a few lucky exceptions). If you want to verify the whitening method in practice, you have plenty of simpler and faster alternatives.
You can download iNMR Passion (freeware) or you can download the freely accessible iNMR (or iNMR reader: they don't allow printing, in demo mode, which is irrelevant in our context). If you have a generic operative system you can ask for a demo copy of MestreNova.
Please don't complain if some of these products are commercial. FTF: where do you publish your results? On your own blog? Second thing: we haven't patented our method and anybody is free to include the algorithm into her/his own program (free or commercial); citing the source would be fair.
A final disclaimer: the whitening method works nicely in 2-D spectroscopy. It's not yet applicable in 3-D cases. When, however, the phase of a 2-D spectrum is impossible to correct manually, it's also impossible to correct it automatically. Obvious!
Automatic phase correction of 2D NMR spectra by a whitening method
Giuseppe Balacco, Carlos Cobas
Published Online: Feb 3 2009 10:57AM
DOI: 10.1002/mrc.2394
If you have followed this blog, you know that many other papers have appeared on a similar subject (automatic phase correction of 1-D spectra). In the past, to discover that the method didn't actually work, you had to write a program by yourself (apart a few lucky exceptions). If you want to verify the whitening method in practice, you have plenty of simpler and faster alternatives.
You can download iNMR Passion (freeware) or you can download the freely accessible iNMR (or iNMR reader: they don't allow printing, in demo mode, which is irrelevant in our context). If you have a generic operative system you can ask for a demo copy of MestreNova.
Please don't complain if some of these products are commercial. FTF: where do you publish your results? On your own blog? Second thing: we haven't patented our method and anybody is free to include the algorithm into her/his own program (free or commercial); citing the source would be fair.
A final disclaimer: the whitening method works nicely in 2-D spectroscopy. It's not yet applicable in 3-D cases. When, however, the phase of a 2-D spectrum is impossible to correct manually, it's also impossible to correct it automatically. Obvious!
Sunday, 1 February 2009


Subscribe to:
Posts (Atom)